6.4 Recombinaison de tableaux
6.4.1 Formats long et large
Le format long d’un tableau de données correspond à un encodage en un minimum de colonnes, les données étant réparties sur un plus grand nombre de lignes en comparaison du format large qui regroupe les données dans plusieurs colonnes successives. Voici un exemple fictif d’un jeu de données au format long :
# # A tibble: 6 x 3
# sex traitment value
# <chr> <chr> <dbl>
# 1 m control 1.2
# 2 f control 3.4
# 3 m test1 4.8
# 4 f test1 3.1
# 5 m test2 0.9
# 6 f test2 1.2Voici maintenant le même jeu de données présenté dans le format large :
# # A tibble: 2 x 4
# sex control test1 test2
# <chr> <dbl> <dbl> <dbl>
# 1 m 1.2 4.8 0.9
# 2 f 3.4 3.1 1.2Dans le format large, les différents niveaux de la variable facteur treatment deviennent autant de colonnes (donc de variables) séparées, et la variable d’origine n’existe plus de manière explicite. Ces deux tableaux contiennent la même information. Bien évidemment, un seul de ces formats est un tableau cas par variables correct. Le format long sera le bon si toutes les mesures sont réalisées sur des individus différents. Par contre, le format large sera correct, si les différentes mesures ont été faites à chaque fois sur les mêmes individus (dans le cas présent, un seul mâle et une seule femelle auraient alors été mesurés dans les trois situations).
C’est la règle qui veut qu’une ligne corresponde à un et un seul individu dans un tableau cas par variables qui permet de décider si le format long ou le format large est celui qui est correctement encodé.
Encoder correctement un tableau de données n’est pas une chose simple. Il peut y avoir plusieurs manières de le représenter. De plus, beaucoup de scientifiques ignorent ou oublient l’importance de bien encoder un tableau sous forme cas par variables. Lorsque vous souhaitez effectuer une représentation graphique, un format peut convenir mieux qu’un autre également, en fonction de ce que vous souhaitez visualiser sur le graphique. Il est donc important de connaître les fonctions permettant de recombiner simplement un tableau de données d’une forme vers l’autre : pivot_longer() et pivot_wider().
L’utilisation des fonction pivot_longer() et
pivot_wider() provenant du package {tidyr} est également
décrite en détails dans R for Data
Science.
Prenons l’exemple d’un jeu de données provenant de l’article scientifique suivant : Paleomicrobiology to investigate copper resistance in bacteria: isolation and description of Cupriavidus necator B9 in the soil of a medieval foundry. L’article est basé sur l’analyse métagénomique de type “shotgun” pour quatre communautés microbiennes (notées c1, c4, c7et c10, respectivement)28. Il en résulte une longue liste de séquences que l’on peut attribuer à des règnes.
shotgun_wide <- tibble(
kingdom = c("Archaea", "Bacteria", "Eukaryota", "Viruses",
"other sequences", "unassigned", "unclassified sequences"),
c1 = c( 98379, 6665903, 81593, 1245, 757, 1320419, 15508),
c4 = c( 217985, 9739134, 101834, 4867, 1406, 2311326, 21572),
c7 = c( 143314, 7103244, 71111, 5181, 907, 1600886, 14423),
c10 = c(272541, 15966053, 150918, 15303, 2688, 3268646, 35024))
rmarkdown::paged_table(shotgun_wide)Ce tableau est clair et lisible… seulement, est-il correctement encodé en cas par variables d’après vous ? Quelle que soit la réponse à cette question, il est toujours possible de passer de ce format large à un format long dans R de la façon suivante :
shotgun_long <- pivot_longer(shotgun_wide, cols = c(c1, c4, c7, c10),
names_to = "batch", values_to = "sequences")
rmarkdown::paged_table(shotgun_long)L’argument cols = est assez flexible. Toutes les formes ci-dessous permettent d’obtenir le même résultat.
shotgun_long <- pivot_longer(shotgun_wide, cols = c1:c10,
names_to = "batch", values_to = "sequences")
shotgun_long <- pivot_longer(shotgun_wide, cols = 2:5,
names_to = "batch", values_to = "sequences")
shotgun_long <- pivot_longer(shotgun_wide, cols = starts_with("c"),
names_to = "batch", values_to = "sequences")Voici la logique derrière pivot_longer(), présentée sous forme d’une animation :
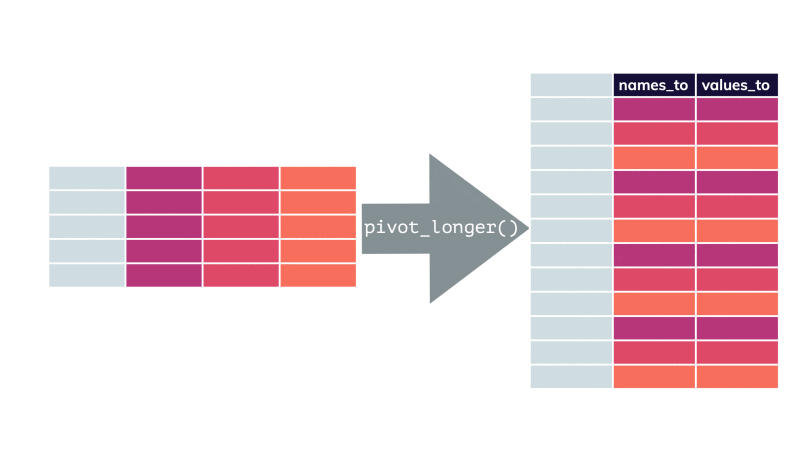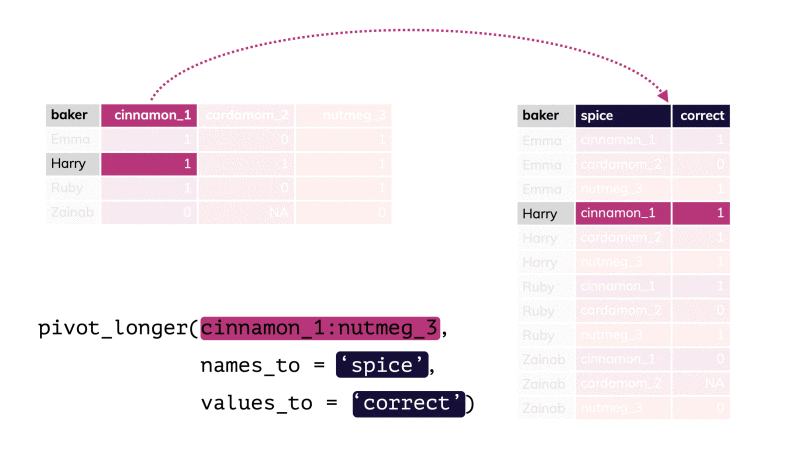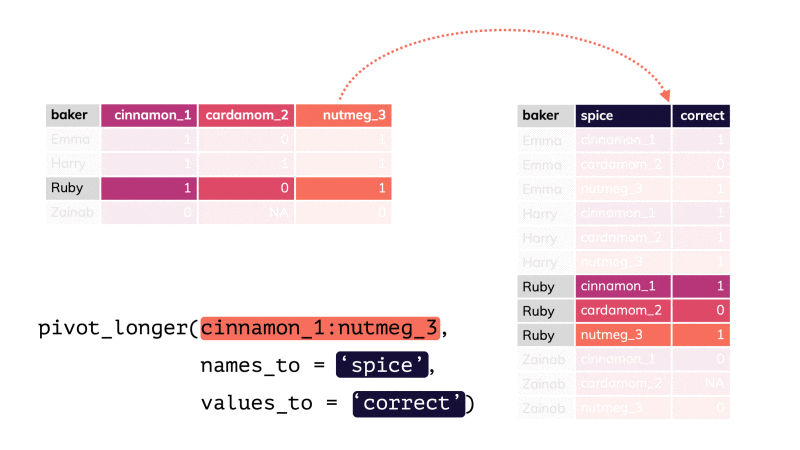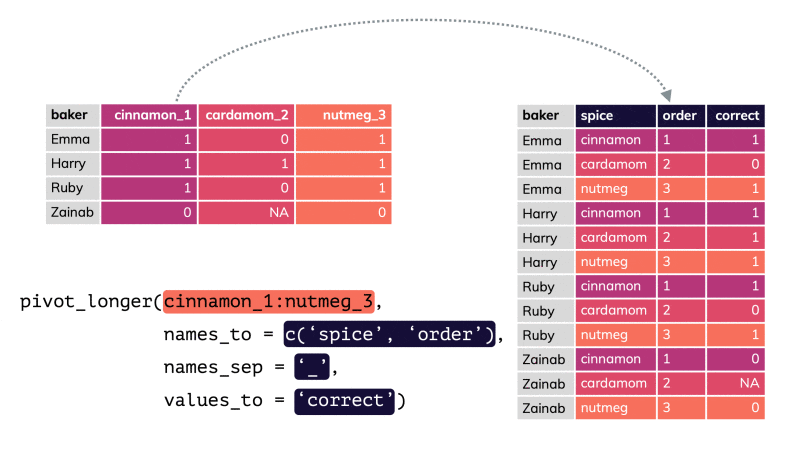
pivot_longer() par apreshill.{kind=link}
Vous conviendrez que le tableau nommé shotgun_long est moins compact et moins aisé à lire comparé à shotgun_wide. C’est une raison qui fait que beaucoup de scientifiques sont tentés d’utiliser le format large alors qu’ici il ne correspond pas à un tableau cas par variables correct, puisqu’il est impossible que les mêmes individus soient présents dans les différents lots (il s’agit de communautés microbiennes indépendantes les unes des autres). De plus, seul le format shotgun_long permet de produire des graphiques pertinents29.
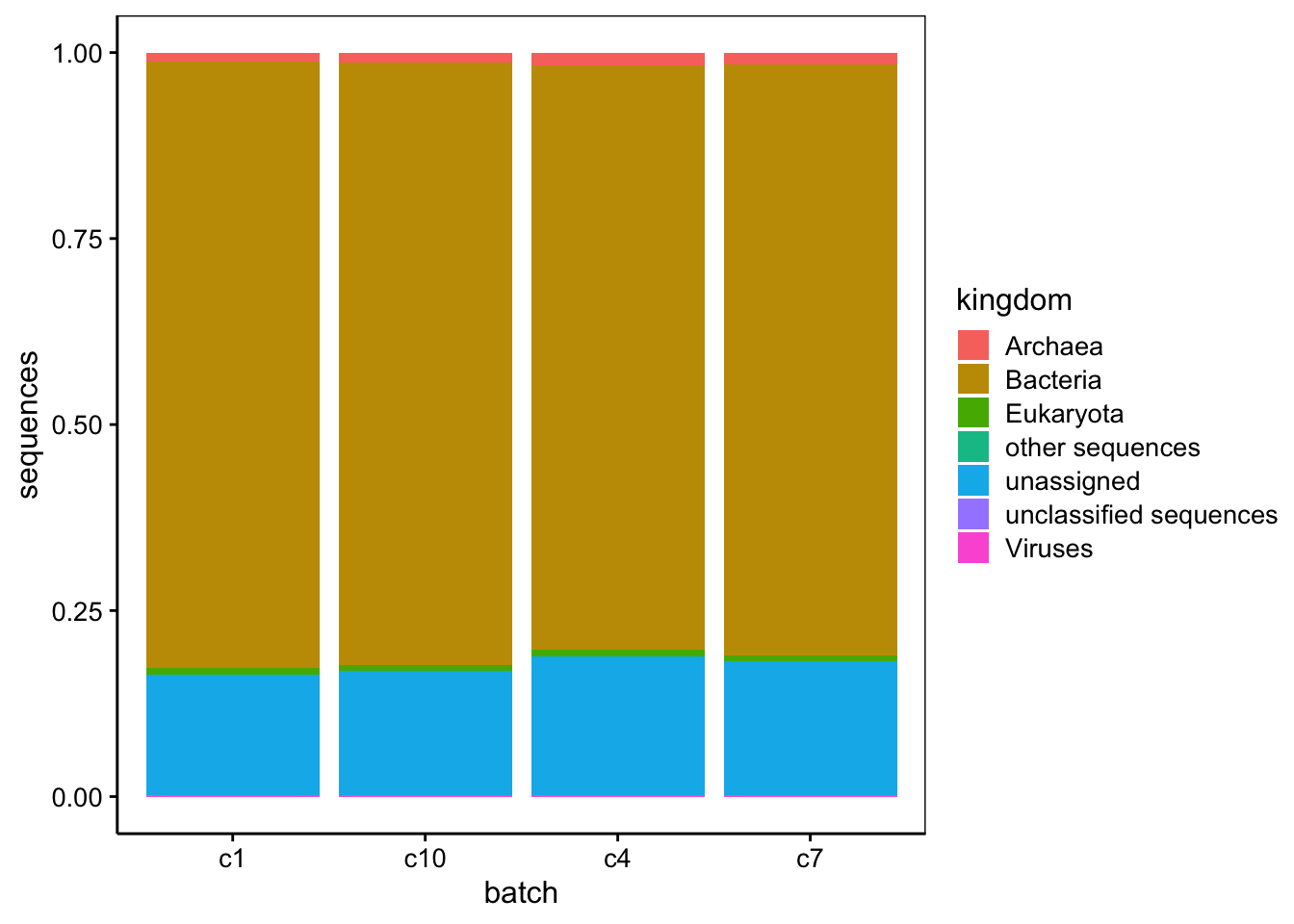
Essayez de réaliser ce type de graphique en partant de shotgun_wide… Bonne chance !
Très souvent, lorsqu’il est impossible de réaliser un graphique avec
chart() ou ggplot() parce que les données se
présentent mal, c’est parce que le jeu de données est encodé de manière
incorrecte ! Si les données sont, par contre, correctement encodées,
demandez-vous alors si le graphique que vous voulez faire est
pertinent.
À vous de jouer !
Pour passer du format long au format large (traitement inverse à pivot_longer()), il faut utiliser la fonction pivot_wider(). Ainsi pour retrouver le tableau d’origine (ou quelque chose de très semblable) à partir de shotgun_long nous utiliserons :
shotgun_wide2 <- pivot_wider(shotgun_long,
names_from = batch, values_from = sequences)
rmarkdown::paged_table(shotgun_wide2)6.4.2 Recombinaison de variables
Parfois, ce sont les variables qui sont encodées de manière inappropriée par rapport aux analyses que vous souhaitez faire. Les fonctions separate() et unite() permettent de séparer une colonne en plusieurs, ou inversement.
L’aide-mémoire Data Import vous rappelle ces fonctions dans sa section Split Cells. Elles sont également décrites en détails dans R for Data Science.
Partons, par exemple, du jeu de données sur la biométrie des crabes du package {MASS} :
La fonction unite() permet de combiner facilement les colonnes sex et species comme montré dans l’exemple ci-dessous. N’hésitez pas à faire appel à la page d’aide de la fonction via ?unite pour vous guider.
La fonction complémentaire à unite() est separate(). Elle permet de séparer une variable en deux ou plusieurs colonnes séparées. Donc, pour retrouver un tableau similaire à celui d’origine, nous pourrons faire :
crabs <- separate(crabs, col = "sp_sex", into = c("sex", "species"), sep = "_")
rmarkdown::paged_table(crabs)À vous de jouer !
Effectuez maintenant les exercices du tutoriel A06La_recombinaison (Recombinaison de tableaux).
BioDataScience1::run("A06La_recombinaison")