5.3 Regroupement avec CAH
À partir du moment où nous avons une matrice de dissimilarités entre toutes les paires d’individus de notre jeu de données, nous pouvons les regrouper en fonction de leurs ressemblances. Deux approches radicalement différentes existent. Soit nous pouvons diviser l’échantillon progressivement jusqu’à obtenir des groupes homogènes (méthodes divisives, telle que les K-moyennes que nous aborderons dans le module suivant), soit nous pouvons regrouper les items semblables petit à petit jusqu’à avoir traité tout l’échantillon (méthodes agglomératives, telle que la classification ascendante hiérarchique étudiée ici).
À vous de jouer !
5.3.1 Dendrogramme
Le dendrogramme est une manière très utile de représenter graphiquement un regroupement. Il s’agit de réaliser un arbre dichotomique ressemblant un peu à un arbre phylogénétique qui est familier aux biologistes (par exemple ici) :
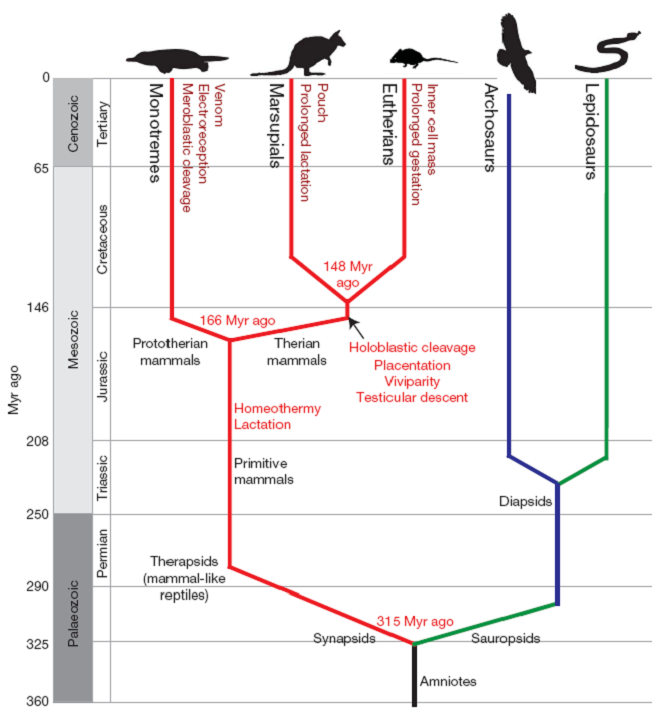
Dans l’arbre, nous représentons des divisions dichotomiques (division d’une branche en deux) matérialisant les divergences au cours de l’évolution. Dans l’arbre présenté ici, l’axe des ordonnées est utilisé pour représenter le temps et les branchements sont placées à une hauteur correspondante en face de l’axe. Le dendrogramme est une représentation similaire de la CAH avec l’axe des ordonnées indiquant à quelle distance le rassemblement se fait.
Dans R, un dendrogramme se construit en deux étapes :
Son calcul par CAH à l’aide de la fonction
cluster(), à partir d’une matrice de dissimilarité dans un objet “dissimilarity”Sa représentation sur un graphique en utilisant
chart().
Partons, pour étayer notre raisonnement, d’une matrice de distances euclidiennes sur les données de zooplancton. Au passage, voyons quelques options utiles pour préparer correctement nos données.
- A la section précédente, nous avons suggéré qu’il peut être utile de standardiser nos données préalablement si les données numériques sont mesurées dans des unités différentes, comme c’est le cas ici. La fonction
scale()ou l’argumentscale = TRUEdedissimilarity()effectuent ce travail. - Limitons, pour l’instant notre ambition à la comparaison de six individus. Afin d’observer tous les cas possibles dans le dendrogramme, nous ne prendrons pas les six premières lignes du tableau, mais les lignes 13 à 18. Cela peut se faire à l’aide de la fonction
slice()que nous n’avons pas encore beaucoup utilisée jusqu’ici. Cette fonction permet de spécifier explicitement les numéros de lignes à conserver, contrairement àfilter()qui applique un test de condition pour décider quelles ligne(s) conserver. Nous pouvons aussi utilise l’argumentsubset =dedissimilarity().
Voici donc notre matrice de distances euclidiennes sur les données ainsi traitées. Les individus initiaux 13 à 18 sont renumérotés 1 à 6. Nous n’imprimons plus ici la matrice de distance obtenue car ce n’est que la première étape du travail vers une représentation plus utile (le dendrogramme).
zoo %>.%
select(., -class) %>.% # Élimination de la colonne class
slice(., 13:18) -> zoo6 # Récupération des lignes 13 à 18
zoo6 %>.% # Matrice de dissimilarité sur données standardisées
dissimilarity(., method = "euclidean", scale = TRUE) -> zoo6std_distNotre objet zoo6std_dist est ensuite utilisé pour calculer notre CAH à l’aide de cluster(). Enfin, chart()se charge de tracer le dendrogramme.
zoo6std_dist %>.%
cluster(.) -> zoo6std_clust # Calcul du dendrogramme
chart(zoo6std_clust) +
ylab("Hauteur")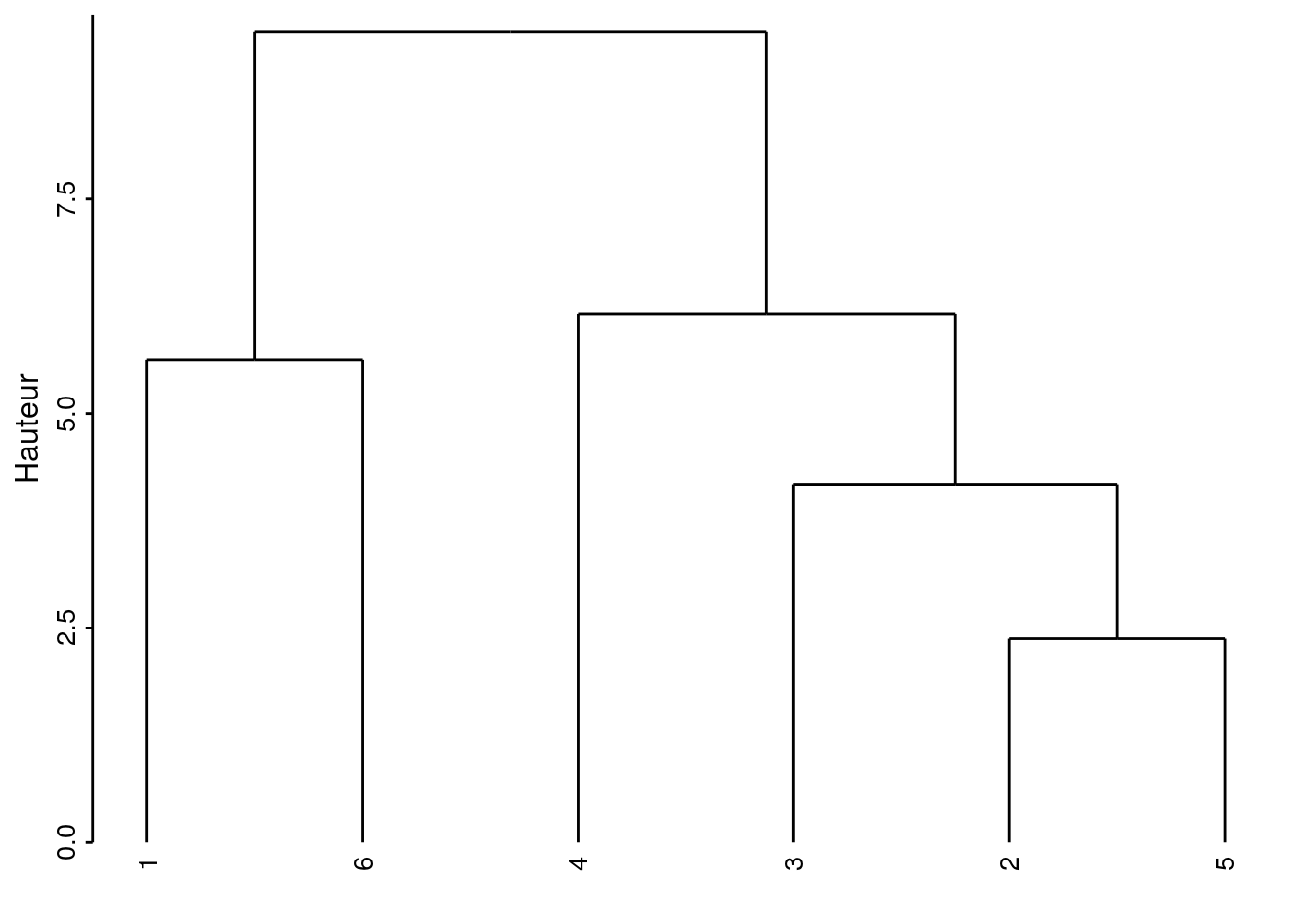
Voici comment interpréter ce graphique. Les deux individus les plus semblables sont le 2 et le 5 (regroupement effectué le plus bas, donc, avec la valeur de l’indice de dissimilarité le plus faible). Ensuite ce groupe formé du 2 et du 5 est le plus similaire au 3. Le 4 se rattache ensuite à ce groupe formé des 2, 3 et 5 mais bien plus haut sur l’axe, indiquant ainsi qu’il s’en différencie un peu plus Enfin, le 1 et le 6 sont rassemblés à un niveau à peu près équivalent. Finalement, le groupe de droite constitué des individus 2, 3, 4 et 5 et celui de gauche contenant le 1 et le 6 sont reliés encore plus haut suggérant ainsi la dissimilitude la plus forte entre ces deux groupes.
Attention : La position des individus selon l’axe horizontal n’est pas importante. Il faut voir le dendrogramme comme un mobile qui peut tourner librement. C’est-à-dire que le groupe constitué de 2, 3, 4 et 5 aurait très bien pu être placé à la gauche de celui constitué de 1 et 6. De même, le sous-groupe 2, 3 et 5 aurait très bien pu être à la gauche du regroupement avec 4 à la droite, etc. Seul compte la hauteur sur l’axe vertical à laquelle les regroupements se font.
Notez que nous n’avons pas utilisé l’information concernant les classes taxonomiques auxquelles les individus appartiennent (nous avons éliminé la variable class en tout début d’analyse). Ce type de regroupement s’appelle une classification non supervisée parce que nous n’imposons pas les groupes que nous souhaitons réaliser13. Cependant, comme nous possédons cette indication du groupe taxonominque de nos six individus par ailleurs dans class, nous pouvons en profiter pour l’utiliser à des fins de vérification :
# [1] Egg_round Poecilostomatoid Poecilostomatoid Decapod
# [5] Calanoid Appendicularian
# 17 Levels: Annelid Appendicularian Calanoid Chaetognath Cirriped ... ProtistLes individus 2, 3 et 5 (“Poecilostomatoid” ou “Calanoid”) sont des copépodes, des crustacés particulièrement abondants dans le zooplancton. Leur forme est similaire. Leur regroupement est logique. L’individu 4 est une larve de décapode, un autre crustacé. Ainsi le regroupement de 2, 3 et 5 avec 4 correspond aux crustacés ici. Enfin, les individus 1 et 6 sont très différents puisqu’il s’agit respectivement d’un œuf rond probablement de poisson et d’un appendiculaire.
5.3.2 Séparer les groupes
Si notre dendrogramme est satisfaisant (nous le déterminons en l’étudiant et en vérifiant que les regroupements obtenus ont un sens biologique par rapport à l’objectif de notre étude), nous concrétisons les groupes que nous souhaitons retenir au final en coupant l’arbre horizontalement à une hauteur choisie et en récupérant les groupes avec predict(). Nous pouvons matérialiser ce niveau de coupure en traçant un trait horizontal rouge dans le dendrogramme en ajoutant geom_dendroline(h = XXX, color = "red"), avec ‘XXX’ la hauteur de coupure souhaitée. Par exemple, si nous coupons l’arbre à une hauteur de 8, nous obtenons deux groupes :
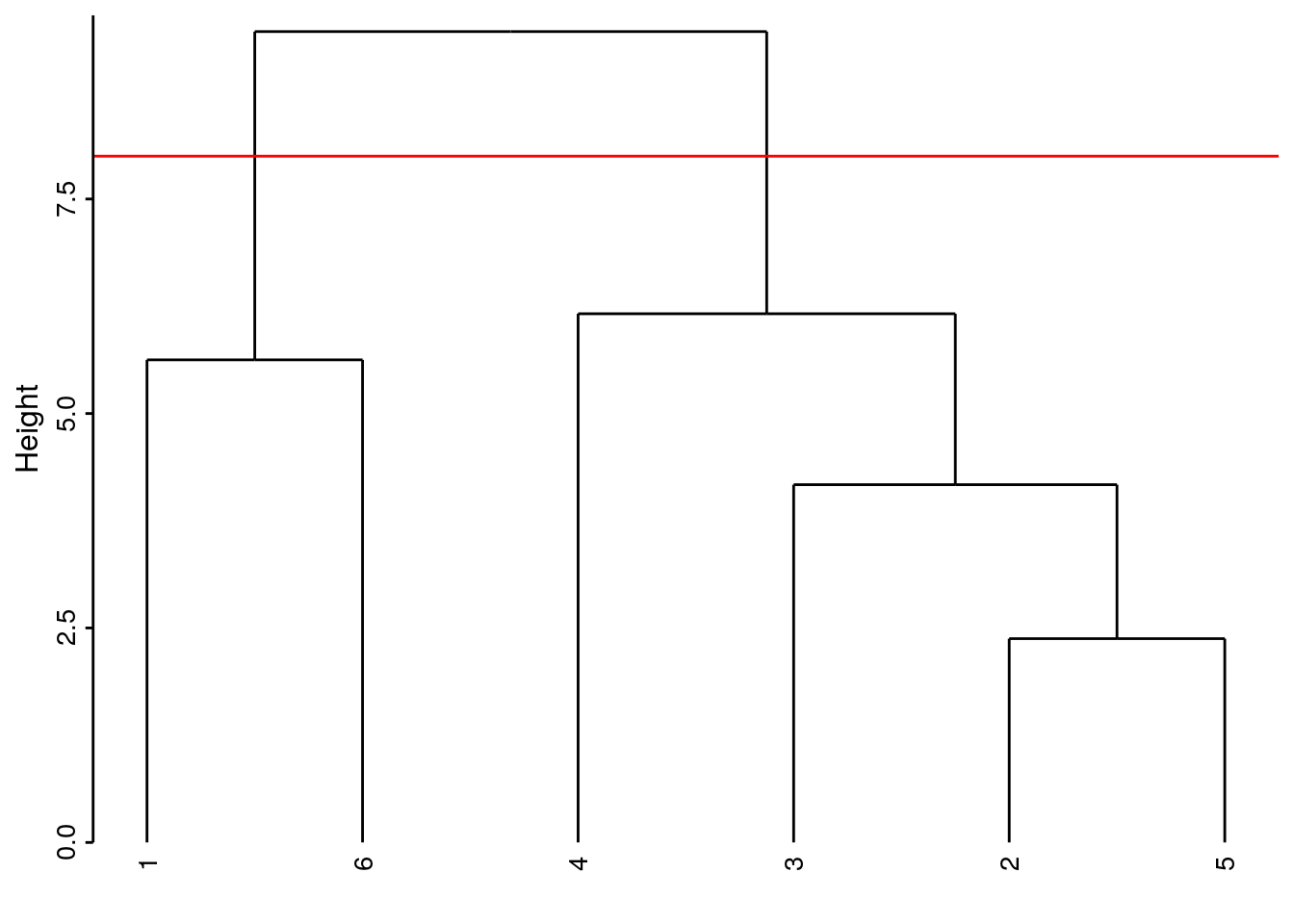
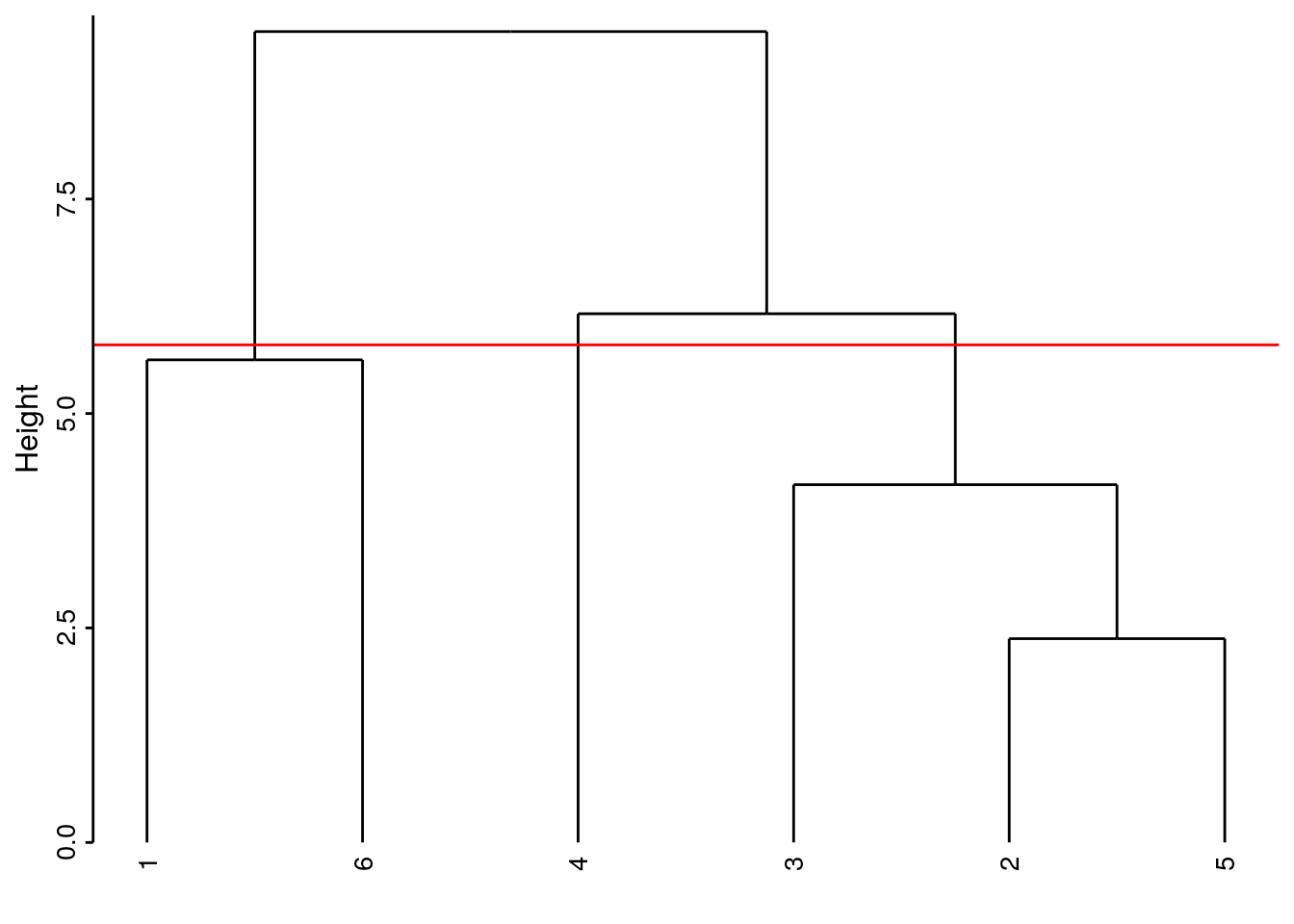
Si nous coupons l’arbre plus bas, par exemple à la hauteur de 5,8, nous obtenons trois groupes :
Les niveaux de coupure ne sont pas toujours très lisibles sur le dendrogramme, mais ils peuvent être obtenus à l’aide de str().
# --[dendrogram w/ 2 branches and 6 members at h = 9.45]
# |--[dendrogram w/ 2 branches and 2 members at h = 5.63]
# | |--leaf "1"
# | `--leaf "6"
# `--[dendrogram w/ 2 branches and 4 members at h = 6.16]
# |--leaf "4"
# `--[dendrogram w/ 2 branches and 3 members at h = 4.17]
# |--leaf "3"
# `--[dendrogram w/ 2 branches and 2 members at h = 2.38]
# |--leaf "2"
# `--leaf "5"À mesure que la hauteur de coupure descend, nous réaliserons quatre, puis cinq groupes. Quel est le meilleur niveau de coupure ? Il n’y en a pas un seul forcément car les différents niveaux correspondent à un point de vue de plus en plus détaillé du regroupement. Néanmoins, lorsque le saut sur l’axe d’un regroupement à l’autre est fort, nous pouvons considérer une séparation des groupes d’autant meilleure. Cela crédibilise d’autant le regroupement choisi. Ainsi la séparation en deux groupes apparaît forte (entre 9.45 et 6.16, sur un intervalle de 3,3 unités donc). De même, la séparation de l’individu 4 par rapport au groupe constitué de 2, 3 et 5 se fait sur un intervalle de 2 unités (entre 6.16 et 4.17). La séparation de l’individu 3 du groupe 2 et 5 est un petit peu moins nette puisqu’elle apparaît aux hauteurs entre 4,17 à 2.38, soit sur un intervalle de 1,8 unités. Ces mesures d’intervalles n’ont aucune valeur absolue. Il faut les considérer uniquement de manière relatives les unes par rapport aux autres, et seulement au sein d’un même dendrogramme.
Ici, deux niveaux de coupure se détachent :
- en deux groupes, nous avons les crustacés séparés des autres.
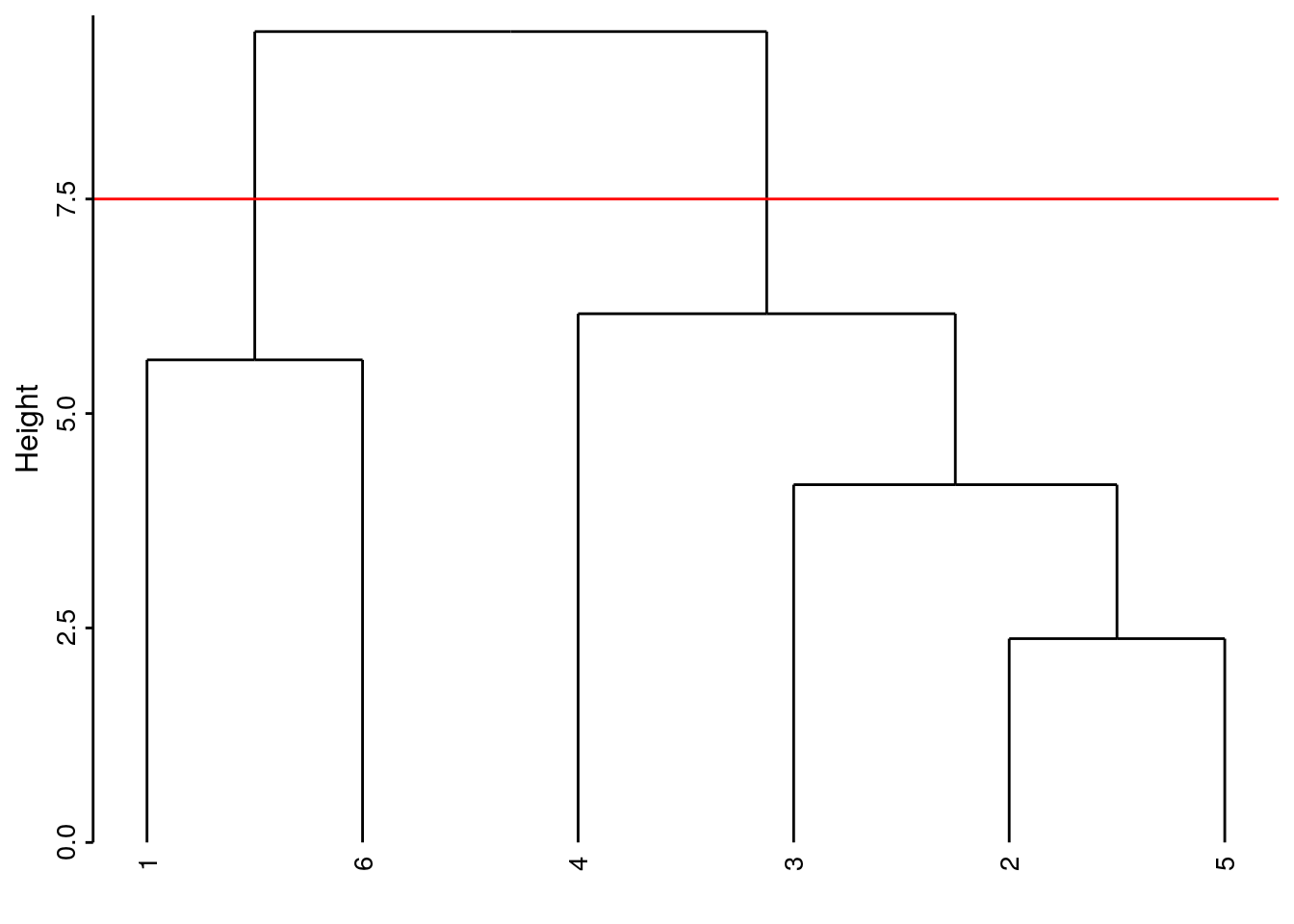
# 1 2 3 4 5 6
# 1 2 2 2 2 1Les individus 1 et 6 sont dans le groupe 1, et les autres dans le groupe 2.
- en quatre groupes, nous avons les copépodes séparés des trois autres items chacun dans un groupe séparé.
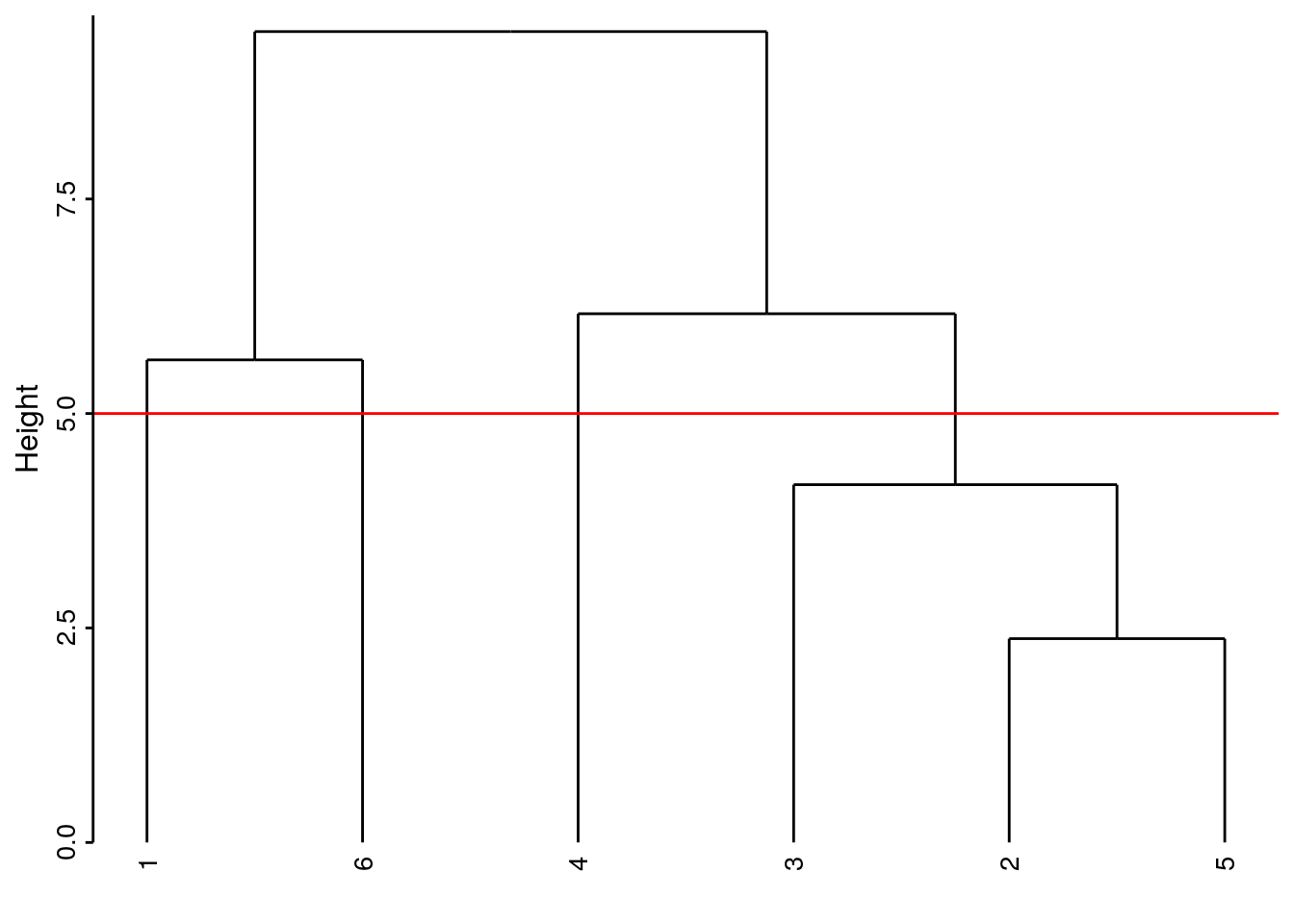
Le groupe des individus 2, 3 et 5 est formé. Les trois autres groupes contiennent des individus uniques 1, 6 et 4 respectivement.
# 1 2 3 4 5 6
# 1 2 2 3 2 4L’individu 1 est dans le groupe 1, les individus 2, 3 et 5 sont dans le groupe 2, l’individu 4 est dans le groupe 3 et enfin l’individu 6 est dans le groupe 4. Donc, predict() numérote ses groupes en fonction du premier item de cette catégorie rencontré dans le tableau des données dans l’ordre dans lequel il se présente.
Nous pouvons ajouter cette variable indiquant les groupes dans le tableau de données à l’aide de la méthode augment() (son nom est .fitted), et utiliser cette information pour en faire d’autres choses utiles. Par exemple, nous représentons un nuage de point à partir de deux variables quantitatives et colorons nos points en fonction de nos groupes comme suit :
zoo6g <- augment(data = zoo6, zoo6std_clust, h = 7.5)
names(zoo6g) # Nom des variables dans ce tableau# [1] "ecd" "area" "perimeter" "feret" "major"
# [6] "minor" "mean" "mode" "min" "max"
# [11] "std_dev" "range" "size" "aspect" "elongation"
# [16] "compactness" "transparency" "circularity" "density" ".fitted"# Nous transformons ces groupes en variable facteur
zoo6g$group <- factor(zoo6g$.fitted)
chart(data = zoo6g, area ~ circularity %col=% group) +
geom_point()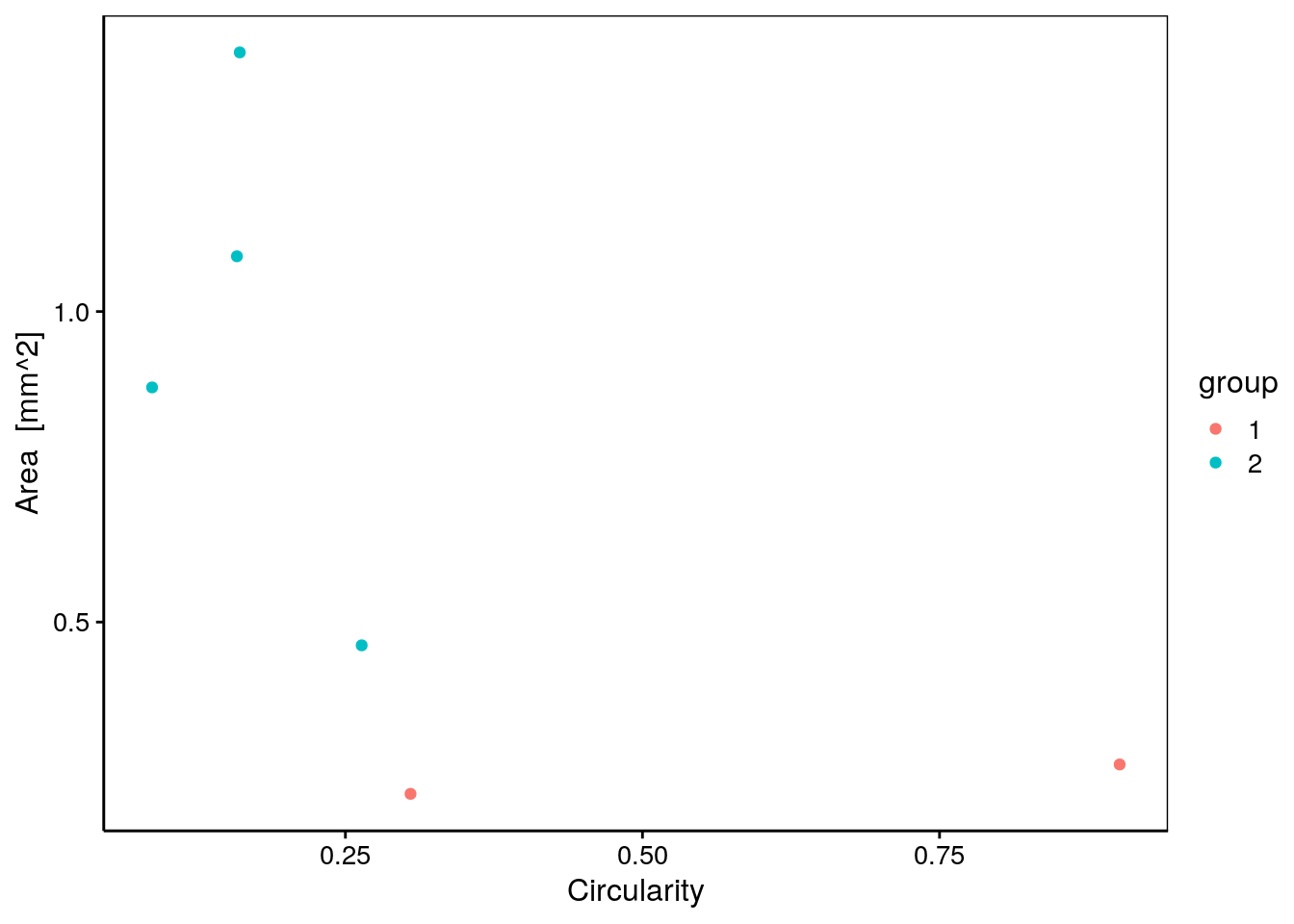
Nous voyons que les copépodes (groupe 2 en turquoise) sont plus grands (area) et moins circulaires (circularity) que les deux autres particules du groupe 1 en rouge. Nous pouvons ainsi réexplorer nos données en fonction de ces groupes pour mieux les comprendre.
5.3.2.1 Méthodes de CAH
Tant que l’on compare des individus isolés entre eux, il n’y a pas d’ambiguïté. Par contre, dès que nous comparons un groupe avec un individu isolé, ou deux groupes entre eux, nous avons plusieurs stratégies possible pour calculer leurs distances (argument method = de cluster()) :
- Liens simples (single linkages en anglais)
cluster(DIS, method = "single"): la distance entre les plus proches voisins au sein des groupes est utilisée.
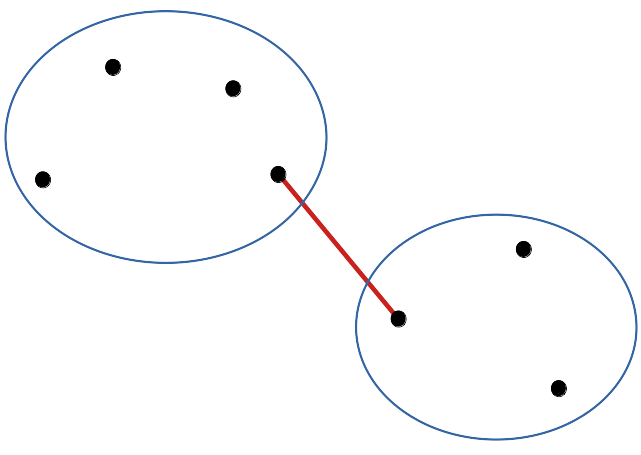
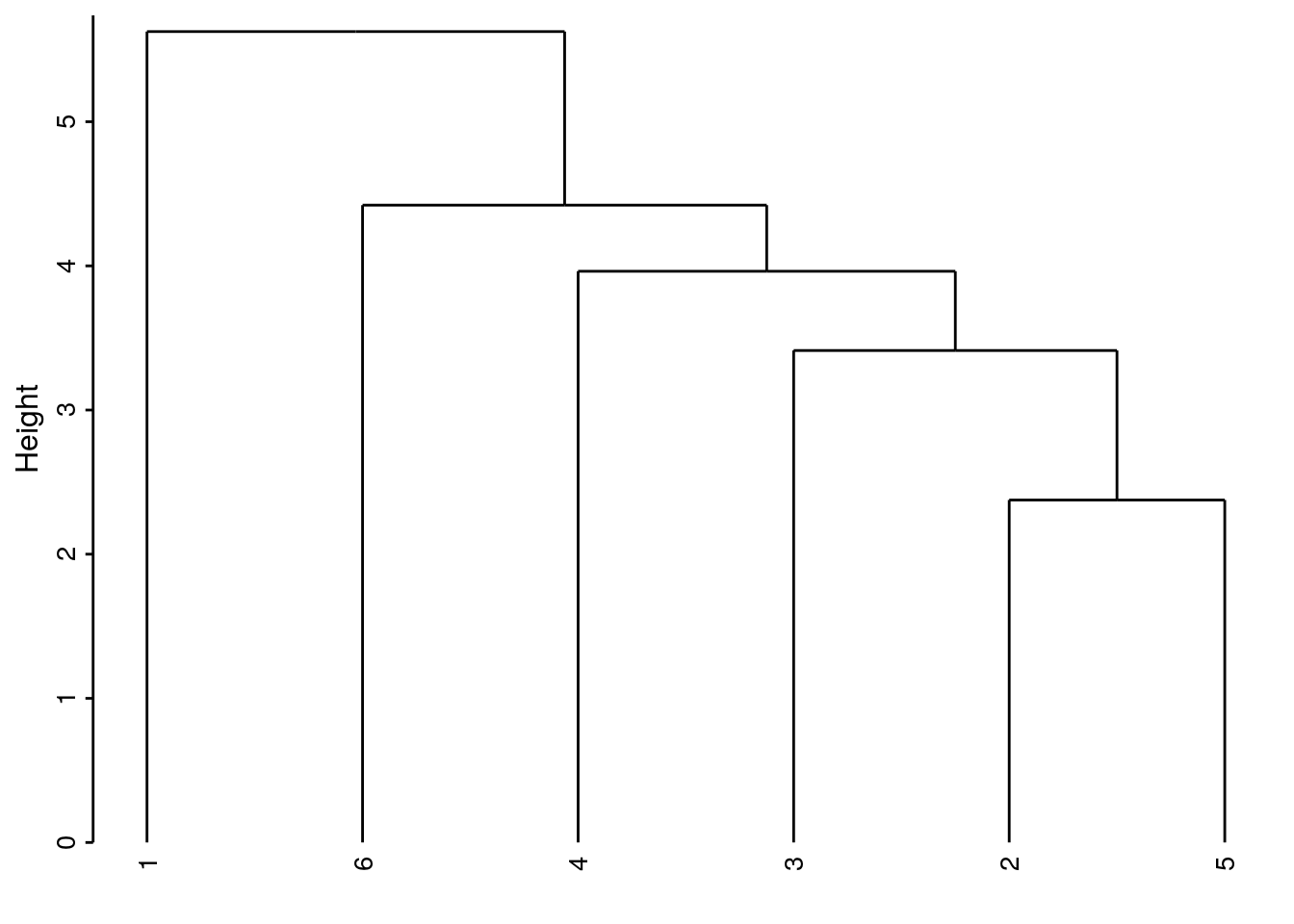
Les données tendent à être agrégées de proche en proche en montrant une gradation d’une extrême à l’autre. Dans le cas d’une variation progressive, par exemple lors d’un passage graduel d’une situation A vers une situation B, le dendrogramme obtenu à l’aide de cette méthode représentera la situation au mieux.
- Liens complets (complete linkages)
cluster(DIS, method = "complete"), méthode utilisée par défaut si non précisée : la distance entre les plus lointains voisins est considérée. C’est le premier dendrogramme que nous réalisé.
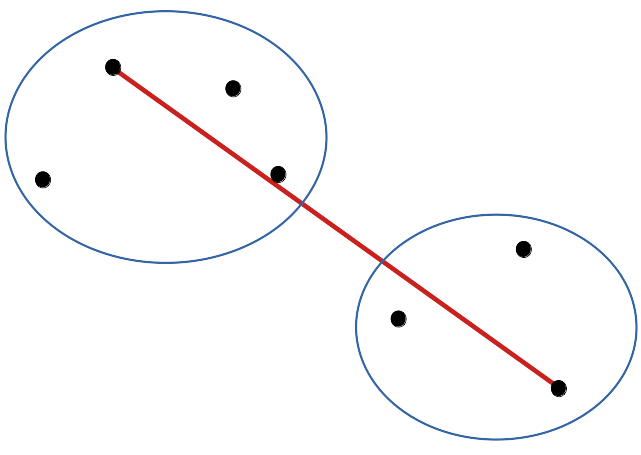
Le dendrogramme a tendance à effectuer des groupes séparés plus nettement les uns des autres qu’avec les liens simples.
- Liens moyens (group-average)
cluster(DIS, method = "average")encore appelée méthode UPGMA : moyenne des liens entre toutes les paires possibles intergroupes.

Nous obtenons une situation intermédiaire entre liens simples et liens moyens.
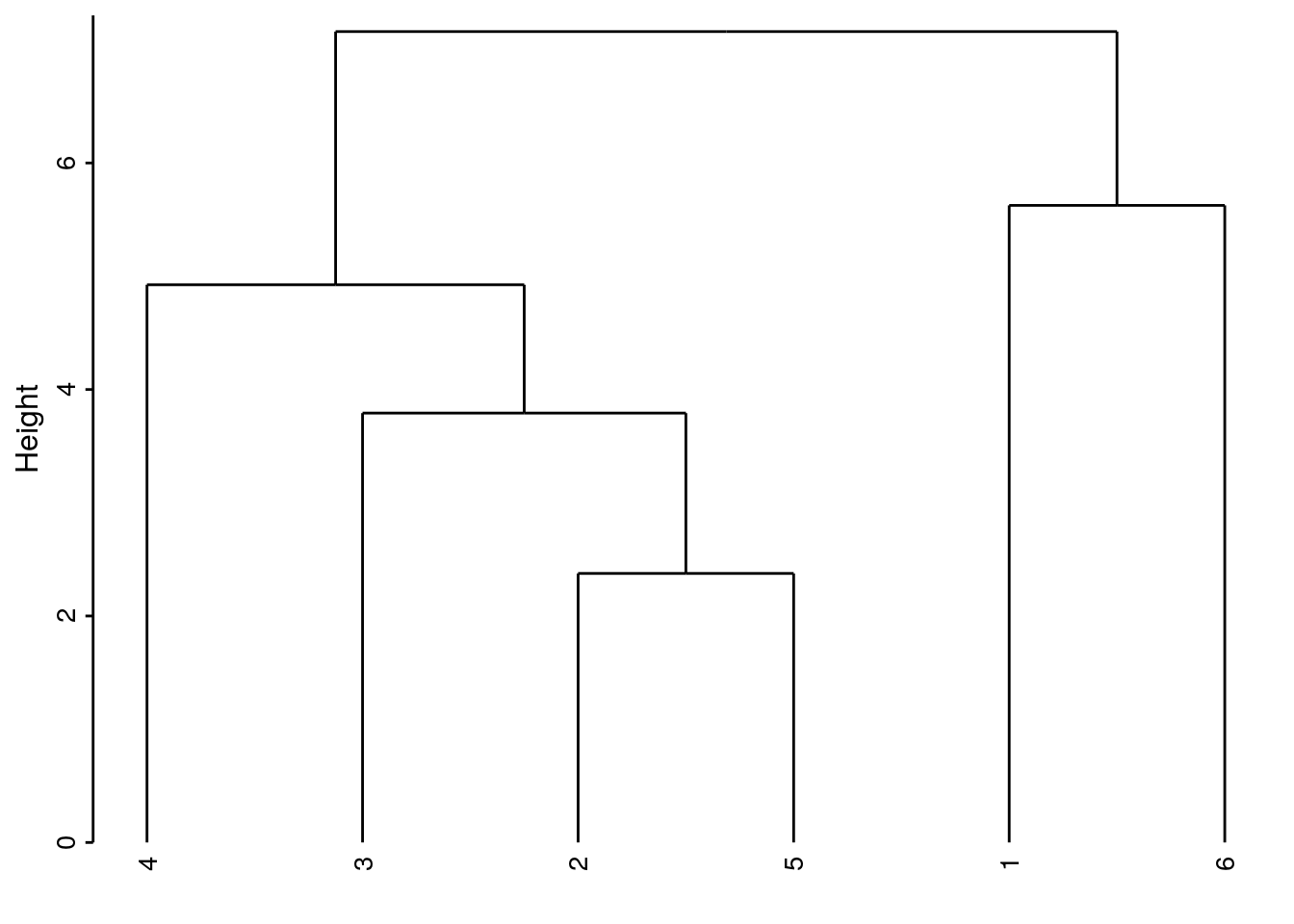
Dans ce cas-ci, le résultat est similaire aux liens simples, mais ce n’est pas forcément le cas tout le temps.
- Méthode de Ward D2
cluster(DIS, method = "ward.D2"). Considérant le partitionnement de la variance totale du nuage de points (on parle aussi de l’inertie du nuage de points) entre variance interclasse et variance intraclasse, la méthode vise à maximiser la variance interclasse et minimiser la variance intraclasse, ce qui revient d’ailleurs au même. Cette technique fonctionne souvent très bien pour obtenir des groupes bien individualisés.
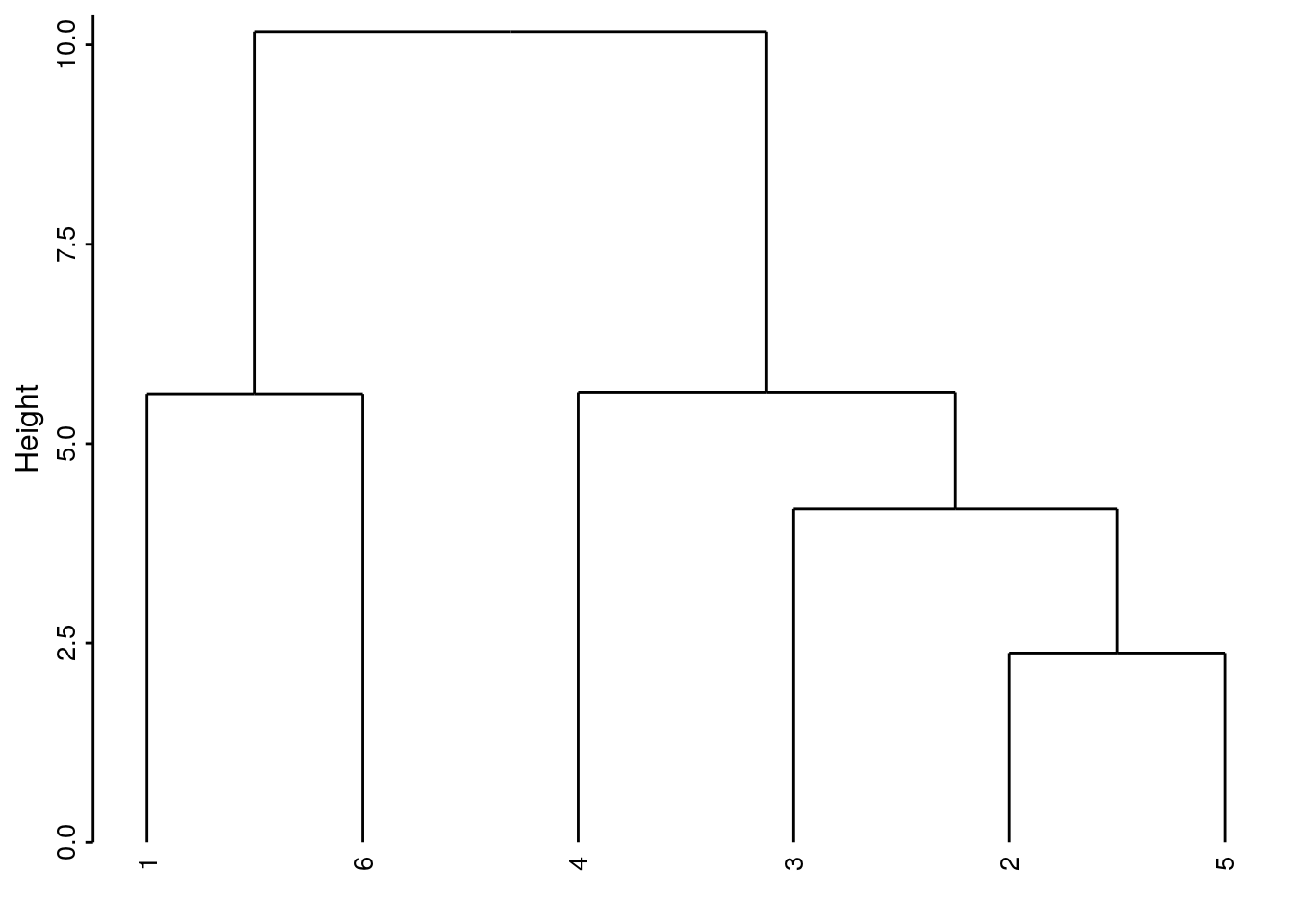
Ici nous obtenons un dendrogramme très proche des liens complets. Encore une fois, ce n’est pas forcément le cas dans toutes les situations.
- Liens médians, centroïdes, …, constituent encore d’autre méthodes possibles, mais moins utilisées. Elles ont l’inconvénient de produire parfois des inversions dans le dendrogramme, c’est-à-dire qu’un regroupement plus avant se fait parfois à une hauteur plus basse sur l’axe des ordonnées, ce qui rend le dendrogramme peu lisible et beaucoup moins esthétique. Dans notre exemple, la méthode centroïde crée une telle inversion à la hauteur de 5 environ sur l’axe des ordonnées entre l’individu 6 et l’individu 1. Alors, qui est regroupé en premier ? Pas facile à déterminer dans ce cas !
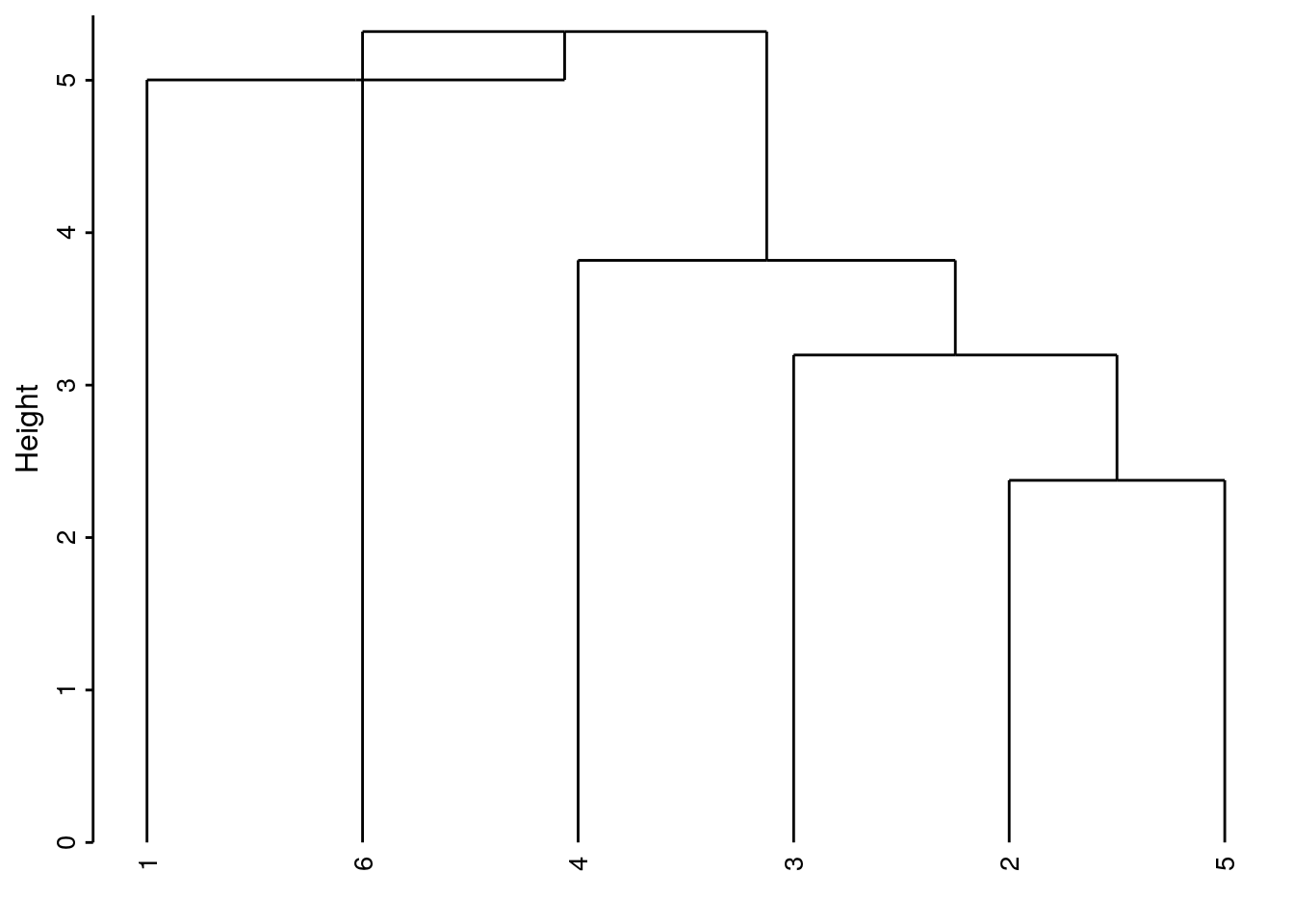
À vous de jouer !
5.3.3 Étude complète
Voici ce que cela donne si nous effectuons une CAH sur le jeu zooplancton complet avec ses 1262 lignes. notez que pour visionner les labels lorsque le nombre d’items augmente, vous pouvez combiner comme ici un dendrogramme horizontal et un étirement en hauteur du graphique en indiquant fig.asp=... comme paramètre du chunk avec une valeur de fig.asp > 1.
zoo %>.%
select(., -class) %>.% # Élimination de la colonne class
# Matrice de dissimilarités sur données standardisées
dissimilarity(., method = "euclidean", scale = TRUE) %>.%
cluster(., method = "ward.D2") -> zoo_clust # CAH avec Ward D2
# Dendrogramme horizontal et sans labels (plus lisible si beaucoup d'items)
chart$horizontal(zoo_clust, labels = FALSE) +
geom_dendroline(h = 70, color = "red") + # Séparation en 3 groupes
ylab("Hauteur")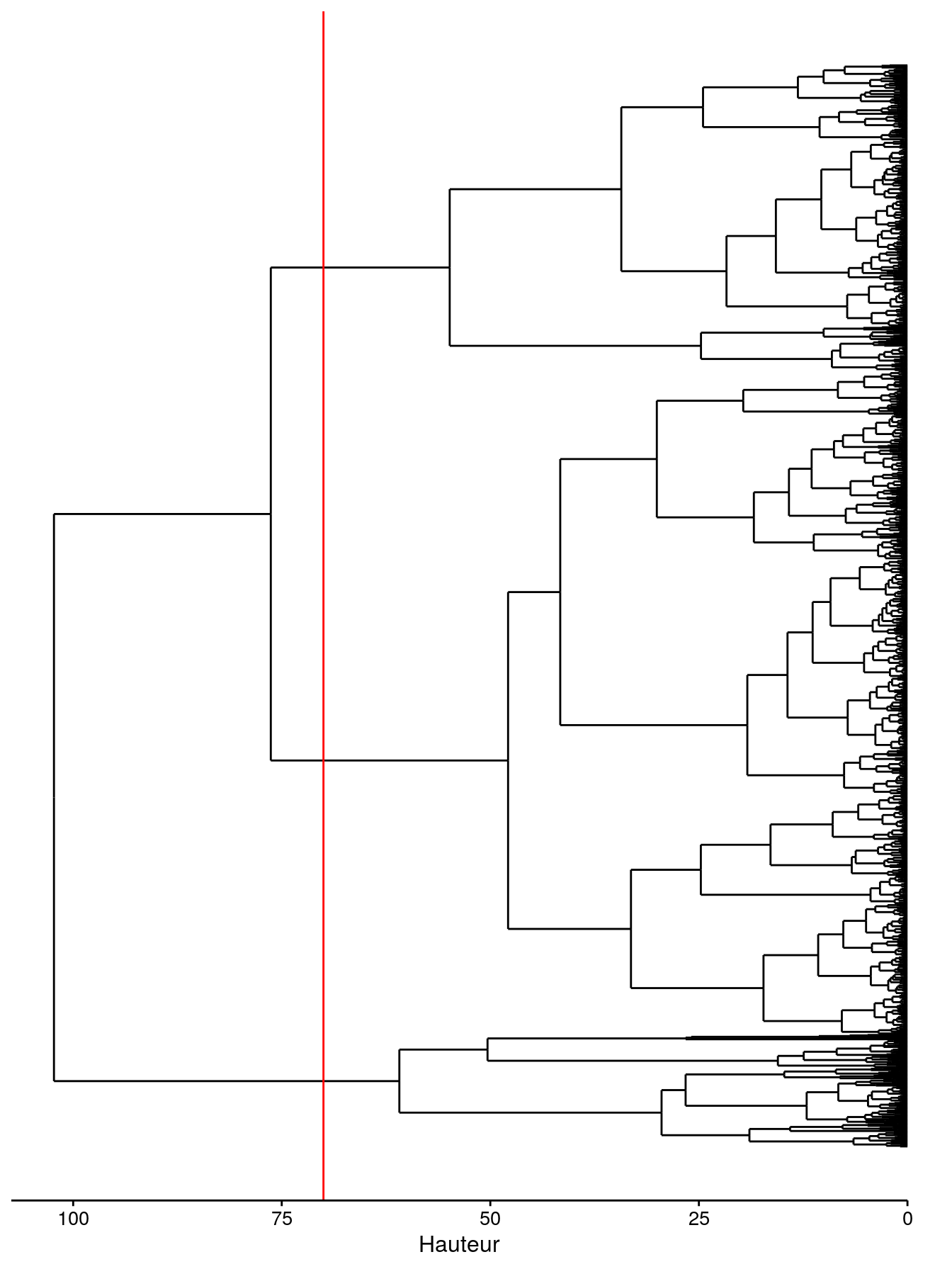
Naturellement, avec 1262 branches, notre dendrogramme est très encombré. Cependant, il reste analysable tant que nous nous intéressons aux regroupements de plus haut niveau (vers le haut du dendrogramme). Notez comme les valeurs sur l’axe des ordonnées ont changé par rapport à nos cas simple à six items (ne jamais comparer les hauteurs entre dendrogrammes différents).
Notre CAH est terminée, nous pouvons récupérer les groupes dans une variable supplémentaire dans le tableau et l’utiliser ensuite.
augment(data = zoo, zoo_clust, h = 70) %>.%
mutate(., group = as.factor(.fitted)) -> zoog
chart(data = zoog, compactness ~ ecd %col=% group) +
geom_point() + # Exploration visuelle des groupes (exemple)
coord_trans(x = "log10", y = "log10") # Axes en log10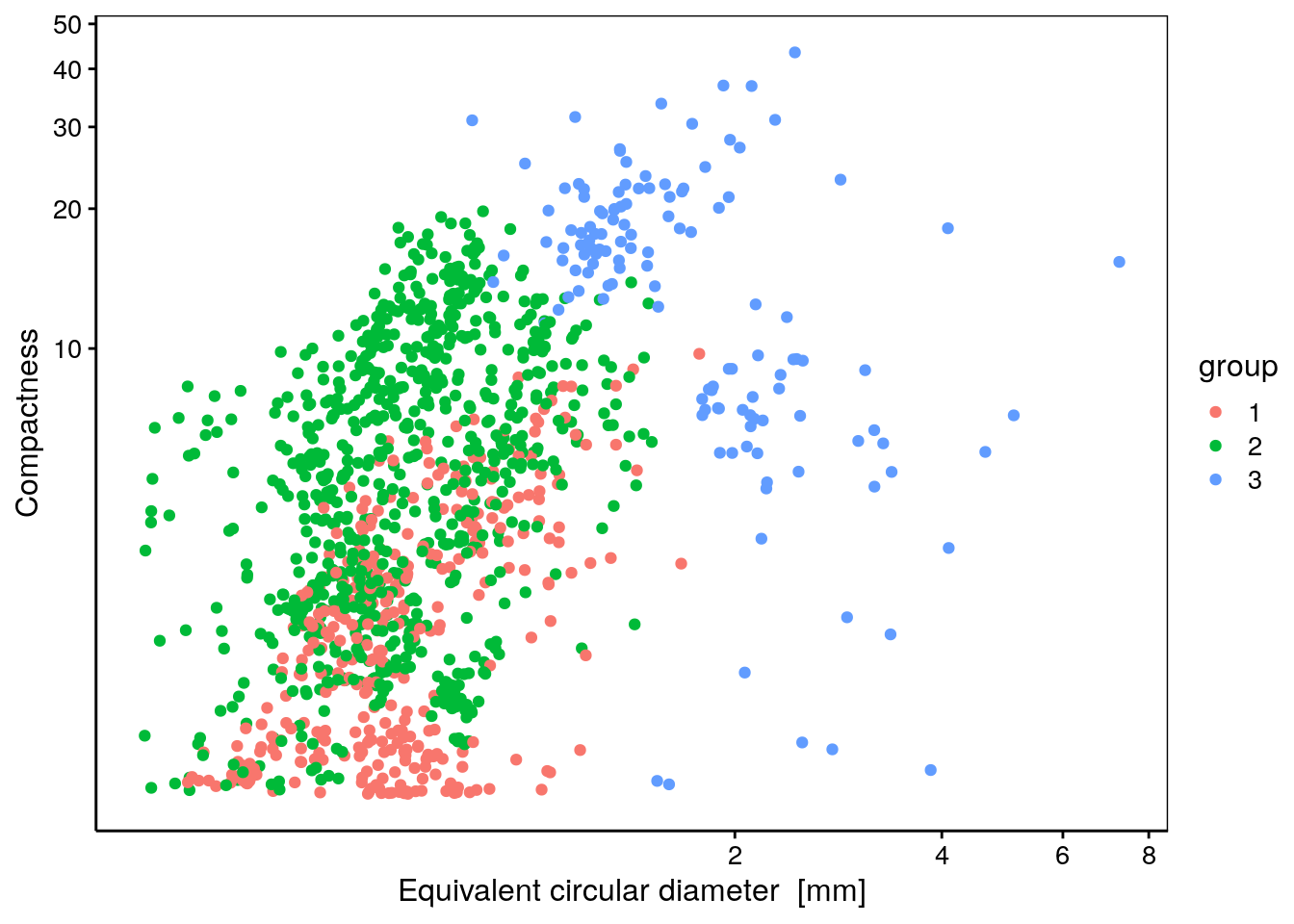
Et de manière optionnelle, si un classement est disponible par ailleurs (c’est le cas ici avec la variable class), nous pouvons réaliser un tableau de contingence à double entrées entre le regroupement obtenu par CAH et le classement obtenu de manière indépendante à cette analyse pour comparaison.
# cah
# classe 1 2 3
# Annelid 31 13 6
# Appendicularian 0 36 0
# Calanoid 9 237 42
# Chaetognath 0 17 34
# Cirriped 1 21 0
# Cladoceran 47 3 0
# Cnidarian 3 5 14
# Cyclopoid 0 50 0
# Decapod 121 5 0
# Egg_elongated 2 48 0
# Egg_round 44 0 5
# Fish 4 46 0
# Gastropod 48 2 0
# Harpacticoid 0 39 0
# Malacostracan 27 66 28
# Poecilostomatoid 20 136 2
# Protist 0 50 0Ainsi, nous pouvons constater que le groupe 1 contient une fraction importante des annélides, des cladocères, des décapodes, des œufs ronds et des gastéropodes. Le groupe 2 contient un maximum des copépodes représentés par les calanoïdes, les cyclopoïdes, les harpacticoïdes et les poecilostomatoïdes. Il contient aussi tous les appendiculaires, tous les protistes, presque tous les œufs allongés, les poissons, et une majorité des malacostracés. Enfin, le groupe 3 contient une majorité des chaetognathes et des cnidaires. Le regroupement a été réalisé uniquement en fonction de mesures effectuées sur les images. Il n’est pas parfait, mais des tendances se dégagent tout de même.
Pour en savoir plus
- La vidéo suivante présente la matière que nous venons d’étudier de manière légèrement différente. Plus d’explications sont également apportées concernant la méthode de Ward.
Une autre explication encore ici.
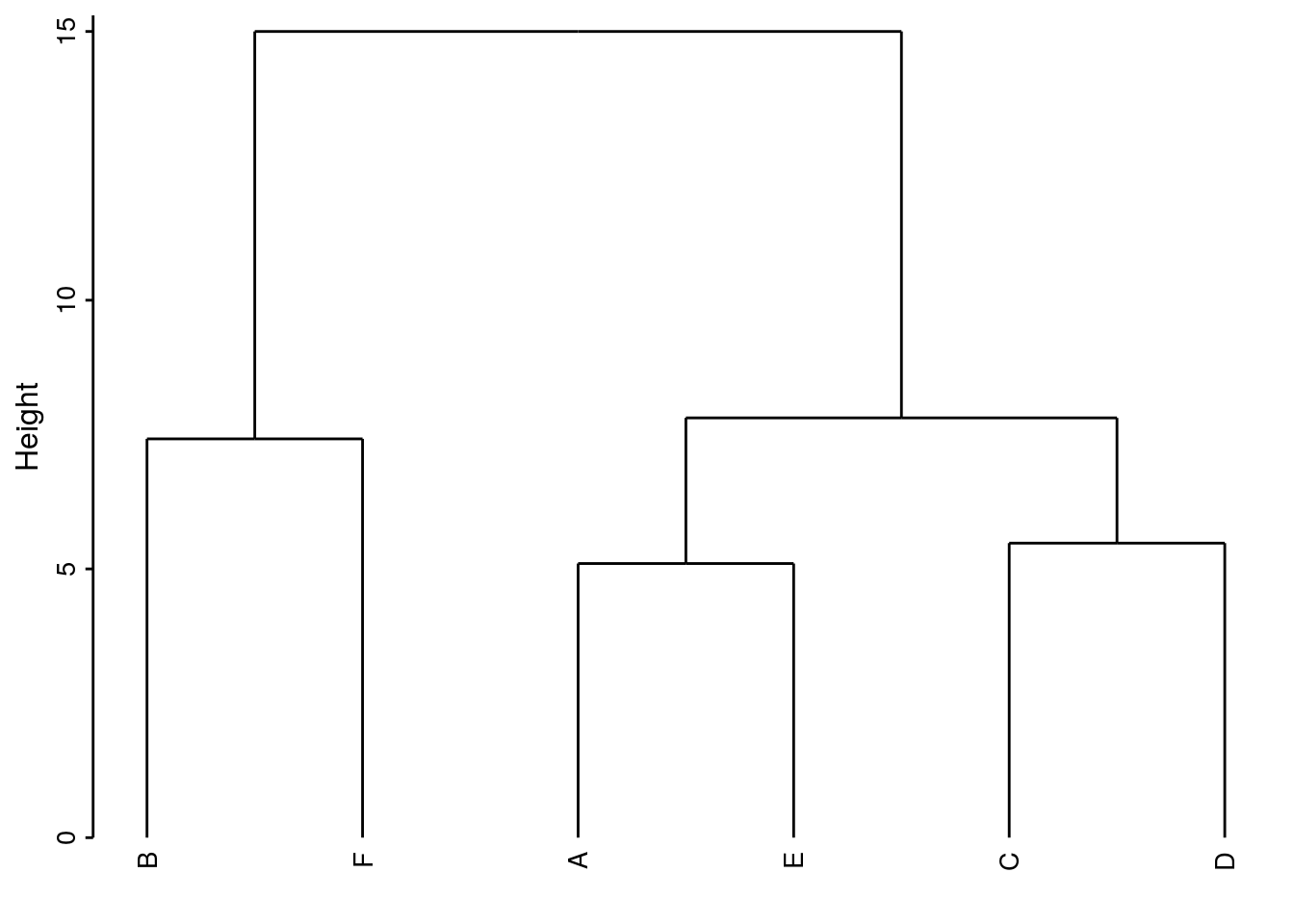
Pour bien comprendre la façon dont une CAH est réalisée, il est utile de détailler le calcul étape par étape sur un exemple simple. Voici une matrice de distances euclidiennes fictive entre 6 stations nommées A à F :
| A | B | C | D | E | |
|---|---|---|---|---|---|
| B | 15 | ||||
| C | 6.4 | 10.86 | |||
| D | 5.2 | 13.04 | 5.48 | ||
| E | 5.1 | 12.37 | 7.28 | 7.81 | |
| F | 10.39 | 7.42 | 5.57 | 9.64 | 9.49 |
Effectuons une CAH manuellement par liens complets et traçons le dendrogramme correspondant.
Etape 1 : Nous repérons dans la matrice de distance la paire qui a l’indice de dissimilarité le plus petit et effectuons un premier regroupement. A_E = groupe I à la distance 5,1. La matrice de distances est simplifiées par rapport à ce groupe I en considérant la règle utilisée (ici, liens complets, donc on garde la plus grande distance entre toutes les paires possibles lorsqu’il y a des groupes).
- Distance entre B et I = B_A = 15
- Distance entre C et I = C_E = 7,28
- Distance entre D et I = D_E = 7,81
- Distance entre F et I = F_A = 10,39
Matrice de distance recalculée :
| I | B | C | D | |
|---|---|---|---|---|
| B | 15.00 | |||
| C | 7.28 | 10.86 | ||
| D | 7.81 | 13.04 | 5.48 | |
| F | 10.39 | 7.42 | 5.57 | 9.64 |
Etape 2 : on répète le processus. C_D = groupe II à la distance la plus petite de 5,48.
- Distance entre I et II = I_D = 7,81
- Distance entre B et II = B_D = 13,04
- Distance entre F et II = F_D = 9,64
Matrice de distance recalculée :
| I | B | II | |
|---|---|---|---|
| B | 15.00 | ||
| II | 7.81 | 13.04 | |
| F | 10.39 | 7.42 | 9.64 |
Etape 3 : B_F = groupe III à la distance 7,42
- Distance entre I et III = I_B = 15
- Distance entre II et III = II_B = 13,04
Matrice de distance recalculée :
| I | III | |
|---|---|---|
| III | 15.00 | |
| II | 7.81 | 13.04 |
Etape 4 : I_II = groupe IV à la distance 7,81
- Distance entre III et IV = III_I = 15
Matrice de distance recalculée :
| III | |
|---|---|
| IV | 15 |
Etape 5 : III - IV = groupe V à la distance 15.
fini !
Voici le dendrogramme résultant :
À vous de jouer !
Effectuez maintenant les exercices du tutoriel B05La_cah (Matrices de distance et classification hiérarchique ascendante).
BioDataScience2::run("B05La_cah")Réalisez l’assignation B05Ia_ligurian_sea.
Si vous êtes un utilisateur non enregistré ou que vous travaillez en dehors d’un cours, faites un “fork” de ce dépôt.
Voyez les explications dans le fichier README.md.
Les techniques complémentaires de classification supervisées seront abordées dans le cours de Science des Données Biologiques III l’an prochain.↩︎